Create a Dataset
On the surface, a dataset in Polaris is simply a tabular collection of data, storing datapoints in a row-wise manner. However, as you try create your own, you'll realize that there is some additional complexity under the hood.
Create a Dataset¶
To create a dataset, you need to instantiate the DatasetV2 class.
from polaris.dataset import DatasetV2, ColumnAnnotation
dataset = DatasetV2(
# Specify metadata on the dataset level
name="tutorial-example",
owner="your-username",
tags=["small-molecules", "predictive", "admet"],
source="https://example.com",
license="CC-BY-4.0",
# Specify metadata on the column level
annotations = {
"Ligand Pose": ColumnAnnotation(
description="The 3D pose of the ligand",
user_attributes={"Object Type": "rdkit.Chem.Mol"},
modality="MOLECULE_3D"
),
"Ligand SMILES": ColumnAnnotation(
description="The 2D graph structure of the ligand, as SMILES",
user_attributes={"Object Type": "str"},
modality="MOLECULE"
),
"Permeability": ColumnAnnotation(
description="MDR1-MDCK efflux ratio (B-A/A-B)",
user_attributes={"Unit": "mL/min/kg"}
)
},
# Specify the actual data
zarr_root_path="path/to/root.zarr",
)
For the rest of this tutorial, we will take a deeper look at the zarr_root_path parameter.
First, some context.
Universal and ML-ready¶
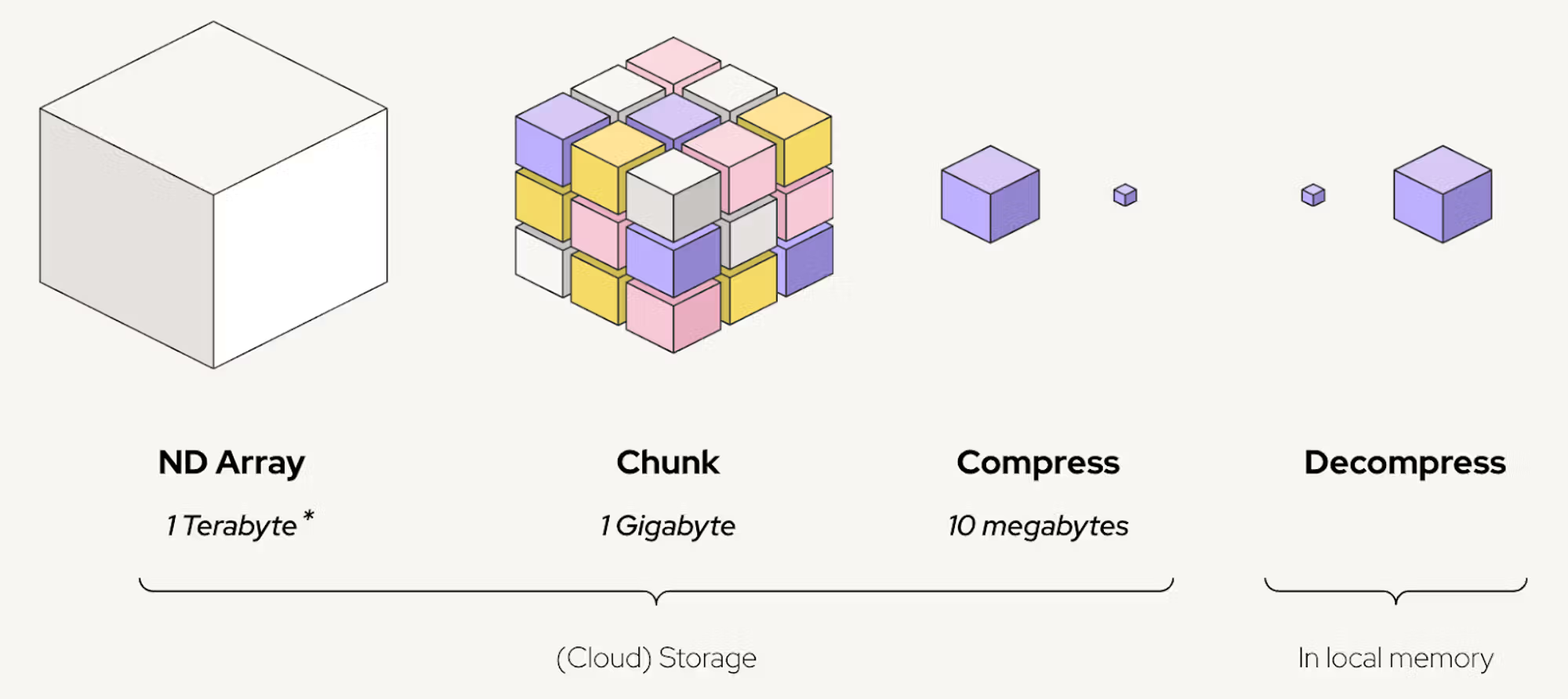 An illustration of Zarr, which is core to Polaris its datamodel
An illustration of Zarr, which is core to Polaris its datamodel
With the Polaris Hub we set out to design a universal data format for ML scientists in drug discovery. Whether you’re working with phenomics, small molecules, or protein structures, you shouldn’t have to spend time learning about domain-specific file formats, APIs, and software tools to be able to run some ML experiments. Beyond modalities, drug discovery datasets also come in different sizes, from kilobytes to terabytes.
We found such a universal data format in Zarr. Zarr is a powerful library for storage of n-dimensional arrays, supporting chunking, compression, and various backends, making it a versatile choice for scientific and large-scale data. It's similar to HDF5, if you're familiar with that.
Want to learn more?
Zarr basics¶
Zarr is well documented and before continuing this tutorial, we recommend you to at least read through the Quickstart.
Converting to Zarr¶
In its most basic form, a Polaris compatible Zarr archive is a single Zarr group (the root) with equal length Zarr arrays for each of the columns in the dataset.
Chances are that your dataset is currently not stored in a Zarr archive. We will show you how to convert a few common formats to a Polaris compatible Zarr archive.
From a Numpy Array¶
The most simple case is if you have your data in a NumPy array.
import numpy as np
data = np.random.random(2048)
import zarr
# Create an empty Zarr group
root = zarr.open(path, "w")
# Populate it with the array
root.array("column_name", data)
From a DataFrame¶
Since Pandas DataFrames can be thought of as labeled NumPy arrays, converting a DataFrame is straight-forward too.
import pandas as pd
df = pd.DataFrame({
"A": np.random.random(2048),
"B": np.random.random(2048)
})
Converting it to Zarr is as simple as creating equally named Zarr Arrays.
import zarr
# Create an empty Zarr group
root = zarr.open(zarr_root_path, "w")
# Populate it with the arrays
for col in set(df.columns):
root.array(col, data=df[col].values)
Things get a little more tricky if you have columns with the object dtype, for example text.
df["C"] = ["test"] * 2048
In that case you need to tell Zarr how to encode the Python object.
import numcodecs
root.array("C", data=df["C"].values, dtype=object, object_codec=numcodecs.VLenUTF8())
# Create an exemplary molecule
mol = Chem.MolFromSmiles('Cc1ccccc1')
mol
from polaris.dataset.zarr.codecs import RDKitMolCodec
# Write it to a Zarr array
root = zarr.open(zarr_root_path, "w")
root.array("molecules", data=[mol] * 100, dtype=object, object_codec=RDKitMolCodec())
A common use case of this is to convert a number of SDF files to a Zarr array.
- Load the SDF files using RDKit to
Chem.Molobjects. - Create a Zarr array with the
RDKitMolCodec. - Store all RDKit objects in the Zarr array.
From Biotite (e.g. mmCIF)¶
Similarly, we can also store entire protein structures, as represented by the Biotite AtomArray class.
from tempfile import TemporaryDirectory
import biotite.database.rcsb as rcsb
from biotite.structure.io import load_structure
# Load an exemplary structure
with TemporaryDirectory() as tmpdir:
path = rcsb.fetch("1l2y", "pdb", tmpdir)
struct = load_structure(path, model=1)
from polaris.dataset.zarr.codecs import AtomArrayCodec
# Write it to a Zarr array
root = zarr.open(zarr_root_path, "w")
root.array("molecules", data=[struct] * 100, dtype=object, object_codec=AtomArrayCodec())
From Images (e.g. PNG)¶
For more convential formats, such as images, codecs likely exist already.
For images for example, these codecs are bundled in imagecodecs, which is an optional dependency of Polaris.
An image is commonly represented as a 3D array (i.e. width x height x channels). It's therefore not needed to use object_codecs here. Instead, we specify the compressor Zarr should use to compress its chunks.
from imagecodecs.numcodecs import Jpeg2k
# You need to explicitly register the codec
numcodecs.register_codec(Jpeg2k)
root = zarr.open(zarr_root_path, "w")
# Array with a single 3 channel image
arr = root.zeros(
"image",
shape=(1, 512, 512, 3),
chunks=(1, 512, 512, 3),
dtype='u1',
compressor=Jpeg2k(level=52, reversible=True),
)
arr[0] = img
Share your dataset¶
Want to share your dataset with the community? Upload it to the Polaris Hub!
dataset.upload_to_hub(owner="your-username")
If you want to upload a new version of your dataset, you can specify its previous version with the parent_artifact_id parameter. Don't forget to add a changelog describing your updates!
dataset.artifact_changelog = "In this version, I added..."
dataset.upload_to_hub(
owner="your-username",
parent_artifact_id="your-username/tutorial-example"
)
Advanced: Optimization¶
In this tutorial, we only briefly touched on the high-level concepts that need to be understood to create a Polaris compatible dataset using Zarr. However, Zarr has a lot more to offer and tweaking the settings can drastically improve storage or data access efficiency.
If you would like to learn more, please see the Zarr documentation.
The End.